Description
PIntron is a novel pipeline for gene-structure prediction based on spliced alignment of transcript sequences (ESTs and mRNAs) against a genomic sequence.
The program is developed and heavily tested on Linux, but it should run also on OS X.
Features
Starting from a genomic sequence and the transcript (EST and/or mRNA) sequences, encoded as (Multi)FASTA files, we predict:
- the exons and introns of the gene(s)
- the full-length isoforms
The output can be easily inspected by the users and can be also easily parsed by simple scripts written in any programming languages. Moreover, the predictions are also reported in the standard GTF format and, thus, can be easily used and visualized by other commonly used software systems (such as the UCSC Genome Browser).
Screenshots
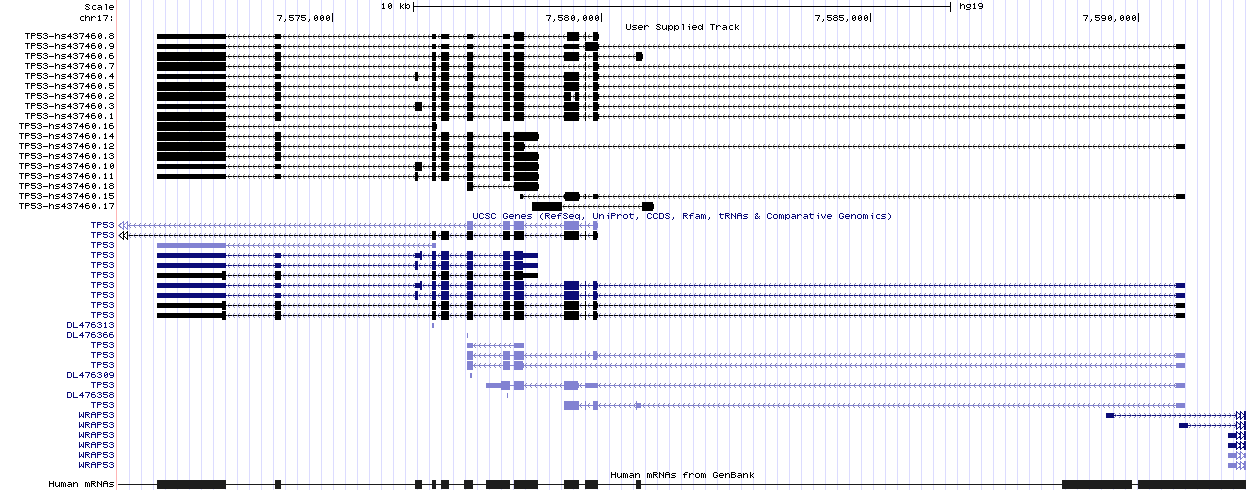
PIntron is a command-line application that can be used on local workstations or on remote servers. Anyway, the computed results can be used with third-party applications to visualize and further process the results.
Here, for example, is the set of full-length isoforms (upper part) computed by PIntron for gene TP53 and visualized by the UCSC Genome Browser.
Main ideas
PIntron is a pipeline composed of five steps:
- Alternative alignments of expressed sequences to a reference genomic sequence are computed and represented in a graph (called embedding graph), using a new ad-hoc fast spliced alignment procedure.
- Biologically meaningful alignments are filtered.
- A draft consensus gene structure is constructed.
- Introns are reconciliated and classified according to general biological criteria.
- A minimal set of full-length isoforms that explains the reconstructed introns and the spliced alignments is finally computed using the method described here.